
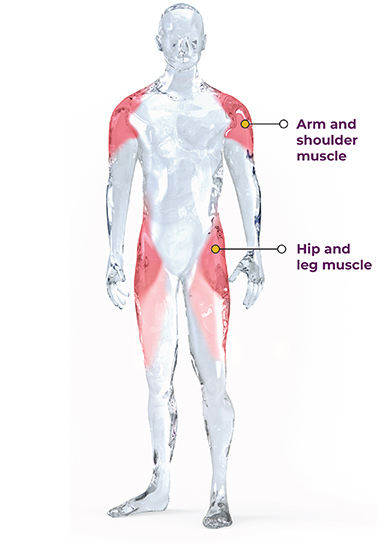
LGMD is a general term for a group of rare genetic disorders which cause weakness and wasting of the muscles specifically around the shoulders, upper arms, hips/ pelvic area, and thighs (limb girdle muscles). The symptoms of LGMD vary depending on the type of the disease, but generally, the initial symptoms include muscle weakness and wasting. Other symptoms may include difficulty with mobility, problems with breathing, and heart abnormalities. It is a progressive disease, and there is no known cure. However, exercise can help improve muscle strength and function, and delay the progression of the disease.
Prevalence of LGMD is estimated to range 1 in 14,500 to 1 in 123,000 people worldwide. However due to the amount of variations to phenotypes and different genetic strands of LGMD, it is estimated to be 1 in 50 000 as an average.
There are two types of LGDM with the distinction between LGMD type 1 and type 2 relating to underlying molecular defect that causes the disease as well whether the disease was inherited through recessive genes or dominant genes. Type 1 referring to dominant gene inheritance and Type 2 referring to recessive gene inheritance. LGMD type 1 is caused by mutations in genes that encode structural proteins of the muscle cell membrane or proteins that are involved in the assembly of the membrane. These mutations result in a disruption of the normal structure and stability of the muscle cell membrane, leading to muscle weakness and wasting. LGMD type 1 is typically associated with earlier onset of symptoms and a more severe disease course compared to LGMD type 2. On the other hand, LGMD type 2 is caused by mutations in genes that encode proteins involved in intracellular signalling pathways or proteins involved in other metabolic processes within muscle cells. These mutations typically result in abnormal muscle fibre function and metabolism, leading to muscle weakness and wasting. LGMD type 2 is generally associated with a later onset of symptoms and a more slowly progressive disease course compared to LGMD type 1. Today, more focus will be placed on LGMD 2i FKRP.
What Specifically is LGMD 2i FKRP?
LGMD Type 2I (also known as LGMD2I) is a type of limb-girdle muscular dystrophy caused by mutations in the FKRP gene and is inherited in an autosomal recessive manner, which means that an affected individual has inherited two copies of the mutated FKRP gene (one from each parent). The FKRP gene provides instructions for making a protein called fukutin-related protein (FKRP). This protein is present in many of the body’s tissues but is particularly abundant in the brain, heart (cardiac) muscle, and muscles used for movement (skeletal muscles). Within cells, FKRP is found in a specialized structure called the Golgi apparatus, where newly produced proteins are modified. FKRP is involved in a process called glycosylation. Through this chemical process, sugar molecules are added to certain proteins. In particular, FKRP adds a molecule called ribitol 5-phosphate to the chain of sugars attached to a protein called alpha (α)-dystroglycan. Glycosylation is critical for the normal function of α-dystroglycan. The α-dystroglycan protein helps anchor the structural framework inside each cell (cytoskeleton) to the lattice of proteins and other molecules outside the cell (extracellular matrix). In skeletal muscles, glycosylated α-dystroglycan helps stabilize and protect muscle fibers. In the brain, it helps direct the movement (migration) of nerve cells (neurons) during early development. The loss of glycosylation of alpha-dystroglycan due to pathogenic variants in FKRP leads to poor linkage of the myocyte to the extracellular matrix, which produces muscle damage as seen in LGMD Type 2i. This ultimately leads to muscle weakness and wasting, particularly in the hip and shoulder muscles. Symptoms typically appear in childhood or adolescence, although the age of onset and severity of symptoms can vary widely.
Age of Onset Question
The age of onset is correlated to the genotype and residual expression of fukutin-related protein. Thus, patients homozygous for the common, mild mutation usually have an onset in the third decade, while patients compound heterozygous for this mutation have onset before age 10, and loose ambulation around age 20 and need assisted ventilation 5-10 years after that. The manifesting symptoms start in the lower extremities with difficulties raising from a chair, climbing stairs and running. Later proximal weakness starts in the arms with visible scapular winging. A third of patients may present with myoglobinuria on physical exertion, before weakness is noticed. Patients may develop a cardiomyopathy that requires medical treatment. In rare cases, a cardiac transplant is required. Left ventricular ejection fraction drops on average 0.4% per year. Patients homozygous for the c.826C>A mutation in FKRP loose ambulation around age 60 and may need assisted ventilation at night, but the timing of these losses is highly variable.
Characteristics
Muscle cramps and myalgia
Most typical presentation is of symmetrical weakness due to scapular-humeral-pelvic weakness, which may be similar to the presentation of Facioscapulohumeral Dystrophy (FSHD), but without facial weakness.
Muscle wasting
Slow progression
Intelligence and cognition is usually normal
Waddling gait – weakness of hip musculature, gait dysfunction, foot drop
Scapular Winging
Joint stiffness
Contractures
Frequent falls
Difficulty walking for longer distances
Loss of ability to perform transfers
Decreased endurance and tolerance to activity
Difficulty climbing stairs and lifting objects
Unable to do overhead activities with progressive difficulty for self-care and movements
Scoliosis / Lordosis
Dysphagia may occur
Gower’s sign may be present in hip muscle weakness
Pseudo-hypertrophy of calf muscles
Cardiomyopathies
Conduction abnormalities, arrhythmias
Late stages may involve difficulty in breathing, respiratory insufficiency
Exercise Considerations for People with LGMD (non-specific)
Exercise can be beneficial for individuals with LGMD, but it is important to consider the specific type and severity of the disease when creating an exercise program. Here are some important considerations for individuals with LGMD:
- Medical Clearance: It is important for individuals with LGMD to obtain medical clearance from a healthcare professional before beginning an exercise program. This will ensure that there are no underlying medical conditions that may be exacerbated by exercise.
- Type of Exercise: Low-impact exercises such as swimming, cycling, and walking are recommended for individuals with LGMD. These exercises are less stressful on the joints and can help maintain muscle strength and mobility.
- Intensity: The intensity of exercise should be adjusted based on the individual’s level of function and ability. Exercise should be performed at a moderate intensity, which is defined as 50-70% of maximum heart rate or having specific RPE scoring of 4-6 / 10.
- Progression: Exercise progression should be gradual to prevent muscle damage or injury. The frequency, duration, and intensity of exercise should be increased gradually over time.
- Rest: Rest is important for individuals with LGMD to prevent muscle fatigue and damage. Adequate rest periods should be included in the exercise program.
- Adaptive Equipment: Individuals with LGMD may require adaptive equipment such as braces or walkers to assist with mobility. These should be incorporated into the exercise program as needed.